
Dr. Altynbek Murat is a quantum computational materials scientist and machine learning expert. His main research interests are in quantum computational materials modeling and materials informatics using machine learning, including the density functional theory (DFT) based ab-initio investigations of novel materials. He works as a Researcher in the Department of Electrical & Computer Engineering at Boston University. His previous research work at King Abdullah University of Science and Technology (KAUST) focused on the advanced computational platform for quantum electron transport in electronic devices and sensors, in collaboration with the Computational Spintronics group at TCD. His PhD research work at Missouri University of Science & Technology was on first-principles investigations of complex oxides as novel transparent conductors.


EXPERTISE
Computational Physics, PhD
- Ab-initio Materials Investigations
- Materials Informatics, Big Data
- Machine Learning, Data Analytics
- High-Performance Supercomputing
Engineering Management, MSc
- Global Project Management
- Leadership & Communications
- Financial Engineering & Investment
Recent Publications
Electronic properties of low-Σ grain boundaries in InAs Altynbek
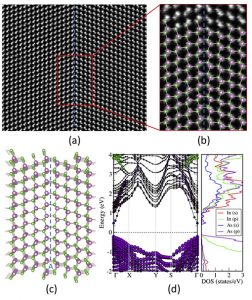 Electronic and structural properties of grain boundaries (GBs) in InAs is investigated. In particular, the energetics and passivation mechanisms of representative low-Σ GBs are studied to establish their relative stability and experimental feasibility. We find that the symmetric-tilt twin-boundary Σ(111) GB is the most stable GB, in excellent agreement with our experimentally characterized GB structures in InAs. Understanding the exact nature of the GB electronic structure and stability is a key step for the further development of InAs based optoelectronic devices. Physical Review Materials, 2, 123604 (2018)
Electronic and structural properties of grain boundaries (GBs) in InAs is investigated. In particular, the energetics and passivation mechanisms of representative low-Σ GBs are studied to establish their relative stability and experimental feasibility. We find that the symmetric-tilt twin-boundary Σ(111) GB is the most stable GB, in excellent agreement with our experimentally characterized GB structures in InAs. Understanding the exact nature of the GB electronic structure and stability is a key step for the further development of InAs based optoelectronic devices. Physical Review Materials, 2, 123604 (2018)Adsorption of the Gas Molecules NH3, NO, NO2, and CO on Borophene
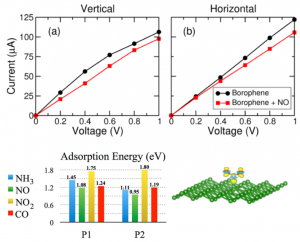 We investigate the adsorption gas molecules NH3, NO, NO2, and CO, on two-dimensional Borophene, which has buckled and line-defective phases. Our results provide a thorough understanding of the interaction between borophene and the gas molecules. An excellent performance of borophene as gas sensor is demonstrated using the nonequilibrium Green’s function method. J. Phys. Chem. C. 122, 26, 14665 (2018)
We investigate the adsorption gas molecules NH3, NO, NO2, and CO, on two-dimensional Borophene, which has buckled and line-defective phases. Our results provide a thorough understanding of the interaction between borophene and the gas molecules. An excellent performance of borophene as gas sensor is demonstrated using the nonequilibrium Green’s function method. J. Phys. Chem. C. 122, 26, 14665 (2018)Quantum Transport Through Tunable Molecular Diodes
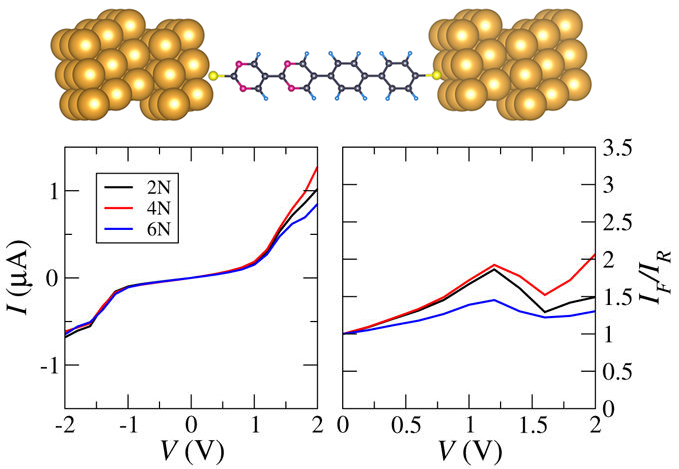 Employing self-interaction corrected density functional theory combined with the non-equilibrium Green’s function method, we study the quantum transport through molecules with different numbers of phenyl (donor) and pyrimidinyl (acceptor) rings in order to evaluate the effects of the molecular composition on the transport properties. Scientific Reports 7, 7324 (2017)
Employing self-interaction corrected density functional theory combined with the non-equilibrium Green’s function method, we study the quantum transport through molecules with different numbers of phenyl (donor) and pyrimidinyl (acceptor) rings in order to evaluate the effects of the molecular composition on the transport properties. Scientific Reports 7, 7324 (2017)Mechanism of H2O induced conductance changes in AuCl4 functionalized CNTs
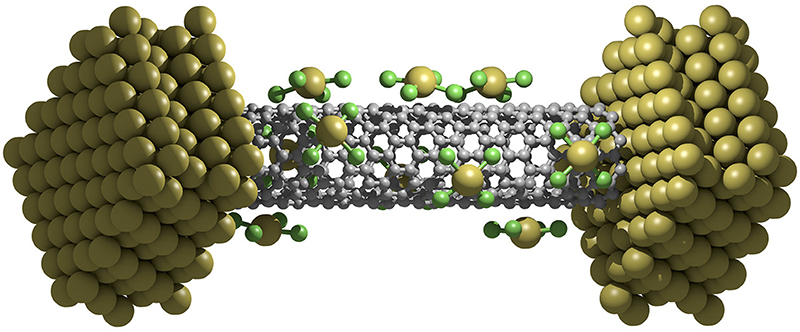 Electronic and quantum transport properties of carbon nanotubes (CNTs) functionalized with AuCl4 molecules are studied using the ab initio self-interaction corrected density functional theory combined with the nonequilibrium Green’s function method. J. Phys. Chem. C. 119, 9568-9573 (2015)
Electronic and quantum transport properties of carbon nanotubes (CNTs) functionalized with AuCl4 molecules are studied using the ab initio self-interaction corrected density functional theory combined with the nonequilibrium Green’s function method. J. Phys. Chem. C. 119, 9568-9573 (2015)
Origin of the p-Type Character of AuCl3 Functionalized Carbon Nanotubes
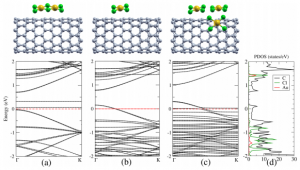 The microscopic origin of the p-type character of AuCl3 functionalized carbon nanotubes (CNTs) is investigated using first-principles self-interaction corrected density functional theory (DFT). The unraveling of the exact nature of the p-doping adsorbates is a key step for further development of AuCl3 functionalized CNTs in water sensor applications.
The microscopic origin of the p-type character of AuCl3 functionalized carbon nanotubes (CNTs) is investigated using first-principles self-interaction corrected density functional theory (DFT). The unraveling of the exact nature of the p-doping adsorbates is a key step for further development of AuCl3 functionalized CNTs in water sensor applications.Carrier Generation in Multicomponent Wide-Bandgap Oxides: InGaZnO4
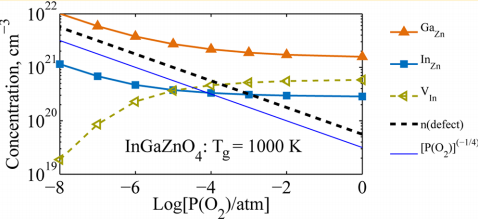 To exploit the full potential of multicomponent wide-bandgap oxides, an in-depth understanding of the complex defect chemistry and of the role played by the constituent oxides is required. In this work, thorough theoretical and experimental investigations are combined in order to explain the carrier generation and transport in crystalline InGaZnO4. J. Am. Chem. Soc. 135, 5685-5692 (2013). [pdf]
To exploit the full potential of multicomponent wide-bandgap oxides, an in-depth understanding of the complex defect chemistry and of the role played by the constituent oxides is required. In this work, thorough theoretical and experimental investigations are combined in order to explain the carrier generation and transport in crystalline InGaZnO4. J. Am. Chem. Soc. 135, 5685-5692 (2013). [pdf]
Electronic properties of layered multicomponent wide-band-gap oxides: A combinatorial approach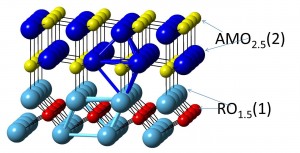
The structural, electronic, and optical properties of 12 multicomponent oxides with layered structure RAMO4, where R3+ = In or Sc, A3+ = Al or Ga, and M2+ = Ca, Cd, Mg, or Zn, are investigated using first-principles density functional approach. Phys. Rev. B 85, 155101 (2012). [pdf]
Composition-dependent oxygen vacancy formation in multicomponent wide-band-gap oxides
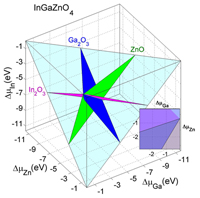 The formation of oxygen vacancy in layered multicomponent InAMO4 oxides and their corresponding binary oxide constituents is investigated using first-principles density functional calculations. Based on the results obtained, we propose several oxides as potential constituents of multicomponent functional materials with tunable properties. Phys. Rev. B 86, 085123 (2012). [pdf]
The formation of oxygen vacancy in layered multicomponent InAMO4 oxides and their corresponding binary oxide constituents is investigated using first-principles density functional calculations. Based on the results obtained, we propose several oxides as potential constituents of multicomponent functional materials with tunable properties. Phys. Rev. B 86, 085123 (2012). [pdf]Research Experience
- Ab-initio Materials Investigations
- Materials Informatics & Data Analysis
- High-Performance Supercomputing
- Global Project Management
- Leadership & Communications
- Financial Engineering & Investment





